Home
driverMAPS is a software to capture positive selection signals using somatic point mutations in cancer.
Overview of the method
We model aggregated exonic somatic mutation counts from many tumor samples (e.g. as obtained from a normal-tumor paired sequencing cohort). Let Yg denote the mutation count data in gene g. We develop models for Yg under three different hypotheses: that the gene is a “non- driver gene” (\(H_0\)), an “oncogene” (\(H_{OG}\)) or a “tumor suppressor gene” (\(H_{TSG}\)). Each model has two parts, a background mutation model (BMM), which models the background mutation process, and a selection mutation model (SMM), which models how selection acts on functional mutations. The BMM parameters are shared by all three hypotheses, reflecting the assumption that background mutation processes are the same for cancer driver and non-driver genes. In contrast the SMM parameters are hypothesis-specific, to capture the different selection pressures in oncogenes vs tumor suppressor genes vs non-driver genes. We fit the hypothesis-specific parameters using training sets of known oncogenes (\(H_{OG}\)), known TSGs (\(H_{TSG}\)), and all other genes (\(H_0\)). (This last set will contain some – as yet unidentified – driver genes, which will tend to make our methods conservative in terms of identifying new driver genes.) To combine information across tumor types we first estimate parameters separately in each tumor type, and then stabilize these estimates using Empirical Bayes shrinkage
Having fit these models, we use them to identify genes whose mutation data are most consistent with the driver genes models (\(H_{OG}\) and \(H_{TSG}\)). Specifically, for each gene g, we measure the overall evidence for g to be a driver gene by the Bayes Factor (likelihood ratio), BFg, defined as: \[BF_g := 0.5 [Pr(Y_g | H_{OG}) + Pr(Y_g | H_{TSG})] / Pr(Y_g | H_0)\] Large values of BFg indicate strong evidence for g being a driver gene, and at any given threshold we can estimate the Bayesian FDR. For results reported here we chose the threshold by requiring FDR<0.1.
Implementation
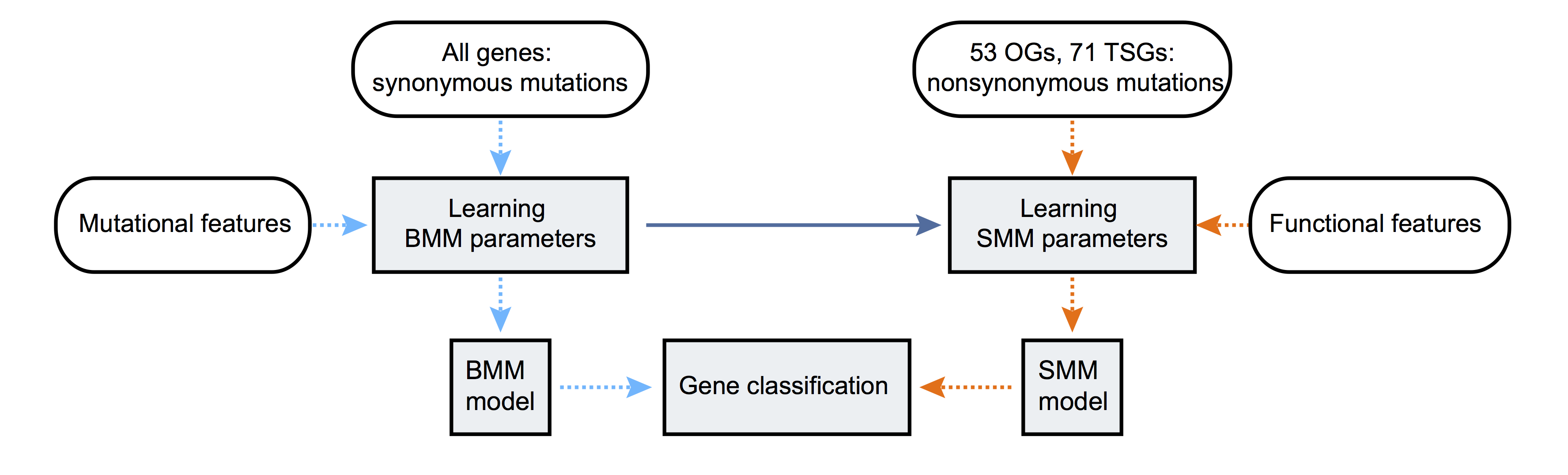
We provided a snakemake package for driverMAPS. This provides all steps in need to produce the results shown in the paper. More specifically here are the step names (as defined in the snakefile file) and their function:
prepdata: This will be first step regardless of the downstream steps.BMRinfer: Learning BMM parameters as shown in the figure above. This step is required regardless of the downstream steps. this will performed for each tumor type independently.Funcvinfer: Part of Learning SMM parameters as shown in the figure above. This step is used when you have collected multiple tumor types and want to reestimate effect sizes for the functional covariates defined in annotation files. If not used, the pre-defined parameters inferred from 20 tumor types used in the paper will be used.ASHfuncv: Part of Learning SMM parameters as shown in the figure above. This step is used when usedFuncvinferas your previous step andASHfuncvwill help to shrink parameters from different tumor types towards mean.HMMinfer: Part of Learning SMM parameters as shown in the figure above. This is used when you want to infer HMM parameters specifically for your tumor types. If not used, the pre-defined parameters inferred from 20 tumor types used in the paper will be used.BayesFactor: gene classificationaddFDR: compute posterior and add FDR.
To install driverMAPS, please see here. In this simplest case, one can use driverMAPS to call drivers from a single tumor cohort (see Quick start). You can also try to re-produce the results showed in the paper by changing the config file (see here), and use filtered mutation lists for 20 tumor types used in driverMAPS paper as input.
Lastest News
2019.09.10 Fixed a missing file bug (“cannot open file ‘data/allgenenames_drivermaps.txt’: No such file or directory”). released v1.0.5.
2019.04.01 Now can be run using a single CPU processor, with 12G memory. The single processor mode takes around 10 hours to finish. To increase speed, we have provided the parallel computing option and the run time can be reduced to 2-3 hours when using 6 cores and 18G memory in total. released v1.0.4.
2018.07.12 Optimized background parameter inference(BMRinfer) procedures. This step now should take < 1 hour.Fixed a bug in getting standard error for parameters. released v1.0.3.
2018.05.12 Added a demo. This demo run can be finished on a laptop computer within half an hour. released v1.0.2
2018.04.18 Bug fix, released v1.0.1
2018.02.01 First version released. 1.0.0
Reference
Zhao, S. et al. Detailed modeling of positive selection improves detection of cancer driver genes. Nat. Commun. 10, 3399 (2019).
Contact
Please send me a message if you have any difficulty running driverMAPS (simingz@uchicago.edu). Thanks!
This R Markdown site was created with workflowr